die den Abbau von zwei Eiweißbausteinen (Phenylalanin und Tyrosin) betrifft und zu einem Anstau des Abbauprodukts Homogentisinsäure (HGA) im Körper führt. Homogentisinsäure ist ein Zwischenprodukt im Abbauvorgang der beiden Eiweißbausteine, den Aminosäuren Phenylalanin und Tyrosin, und wird durch ein Enzym mit dem Namen Homogentisinsäureoxidase abgebaut. Ein Enzymdefekt führt zu einer nicht vorgesehenen Anreicherung der HGA in den Körperflüssigkeiten. Ein Teil der HGA wird mit dem Urin ausgeschieden und oxidiert unter Sauerstoff. Dieser Vorgang ist für die Dunkelfärbung des Urins verantwortlich. Nicht ausgeschiedene HGA lagert sich im Bindegewebe und Knorpelgewebe des Körpers ab.
Gelegentlich werden Neugeborene aufgrund der dunkel gefärbten Windeln diagnostiziert. Viele Betroffene sind symptomlos und bemerken bis in das Erwachsenenalter hinein ihre Krankheit nicht. Nach dem dritten Lebensjahrzehnt wird eine ungewöhnliche Pigmentierung von über Knorpel liegender Haut gesehen. Beschwerden seitens der Muskulatur und des Skeletts beginnen in der 3. Dekade mit Steifheit und Rückenschmerzen. Die Beteiligung der großen peripheren Gelenke setzt meist mehrere Jahre nach Beginn der Beschwerden an der Wirbelsäule ein. Oft geht die Gelenkfunktion vollständig verloren und ein chirurgischer Ersatz wird erforderlich. Die ochronotische Arthopathie (Gelenkerkrankung) ist meist ein degenerativer Prozess, aber bei einigen Fällen können auch Gelenkentzündungen gesehen werden. Die Gelenkbeweglichkeit ist eingeschränkt, Ankylosen (vollständige Gelenksteife) sind möglich.
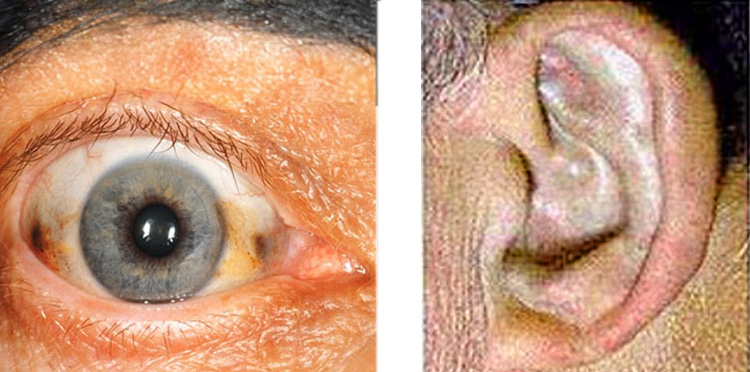
Zu den typischen Alkaptonurie -Symptomen gehören bei den meisten Patienten im Laufe der Zeit blau-schwarz gefärbte Knorpel der Ohren. Ursache ist (wie bei allen folgenden Symptomen) die oxidierte Homogentisinsäure (HGA), die sich dort ansammelt. Dieser Vorgang ist nicht mit Schmerzen verbunden.
Fast alle Betroffenen haben mit der Zeit dunkle Flecken in der weißen Augenhaut. Diese Verfärbungen sind ungefährlich, es treten keine Sehbehinderungen auf.
Nahezu alle Patienten berichten über ihren dunklen Urin. Er ist nahezu schwarz wie Cola, weil die Homogentisinsäure an der Luft oxidiert.
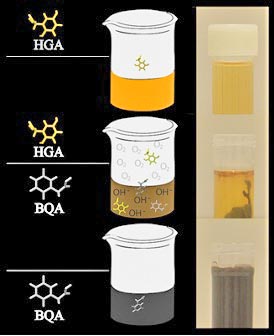
Verfärbung des Urins durch Oxidation von HGA zu BQA
Auch Muskelschmerzen sind ein häufig auftretendes Symptom der Alkaptonurie. Leichte Schmerzen verbunden mit Steifigkeit erleben fast alle Patienten im Laufe ihrer Krankheit.
Zu den größeren Problemen, die jeder AKU-Betroffene mit der Zeit entwickelt, gehören Gelenkprobleme. Im vierten Lebensjahrzehnt treten wohl bei allen Rückenschmerzen auf. Anschließend folgen die Knie, Hüften, Schultern und auch Kleingelenke. Umfang und Intensität der Schmerzen und Bewegungseinschränkungen sind von Patient zu Patient verschieden. Ursache ist wie immer die Anreicherung von HGA im Knorpelgewebe, die dann im weiteren Ablauf zur Ochronose mit der typischen Dunkelfärbung der Knorpel und des Gelenkes führt. Der Knorpel ist dann brüchig und kann seine Aufgaben nicht mehr ausreichend erfüllen. Die Folge ist meist Arthrose. Nahezu alle AKU-Betroffene haben im höheren Lebensalter mindestens ein künstliches Gelenk. Häufig bleibt es nicht das einzige. Ansonsten werden Schmerzen mit Schmerzmedikamenten, Physiotherapie und angepasster Lebensweise in Schach gehalten.
Das folgenschwerste Problem sind Komplikationen mit dem Herzen aufgrund von AKU. HGA kann sich in den Arterien und Herzklappen absetzen. Die Homogentisinsäure führt dann zu brüchigen und verengten Blutgefäßen. Ein Herzklappenersatz ist dabei nicht selten notwendig.
Weitere Symptome können Nieren-, Blasen- und Prostatasteine sein. Auch die Zähne, das zentrale Nervensystem und der Knorpel des Kehlkopfs, Hautverfärbungen und andere Systeme können betroffen sein.
Insgesamt bleibt festzuhalten, dass bei der Mehrzahl der Betroffenen – bei angepasstem Verhalten – die Krankheit nicht zu großen Einschränkungen der Lebensqualität führt oder sich die Lebenserwartung verkürzt. Im Alter kann sich der Gesundheitszustand auch erheblich verschlechtern , so dass der Alltag sehr beschwerlich wird und man gegebenenfalls auf einen Rollator angewiesen ist.
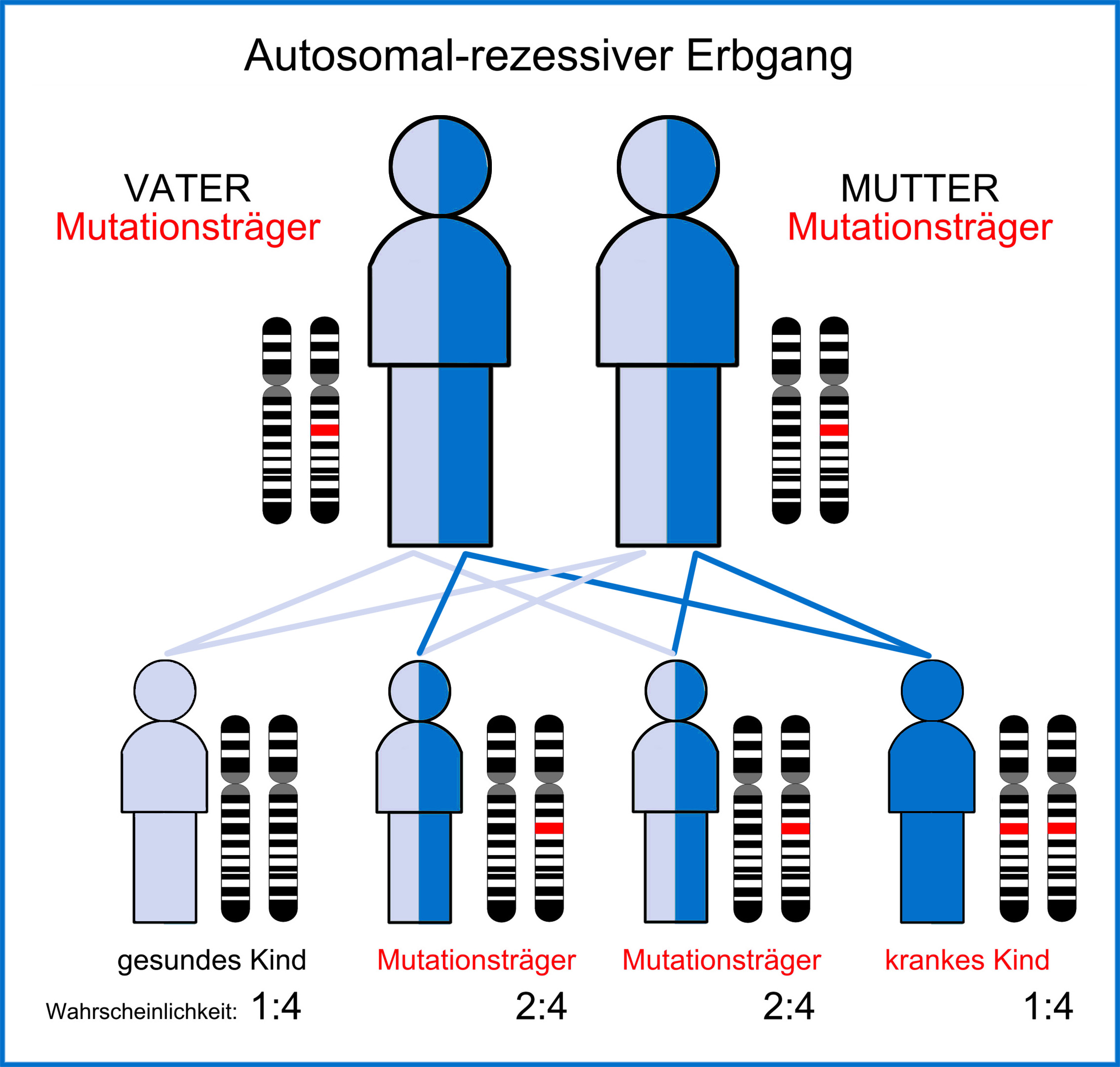
AKU wird autosomal rezessiv vererbt und ist daher angeboren. Typischerweise tragen beide Elternteile ein gesundes und ein erkranktes Gen in sich. Das gesunde Gen dominiert in diesem Falle, so dass klassischerweise beide Elternteile überhaupt keine Symptome der Erkrankung haben. Bei den Kindern ist es nur dann der Fall, wenn beide Elternteile das jeweils kranke Gen an das Kind weitergeben, daraufhin kommt es bei dem Kind zu AKU. Dies ist rein statistisch nur für jedes 4. Kind aus einer solchen Verbindung der Fall. Die Wahrscheinlichkeit an Alkaptonurie zu erkranken kann auch Generationen überspringen, wenn das erkrankte Gen weitergegeben worden ist.
Die Häufigkeit ist sehr unterschiedlich verteilt mit 1:100.000-250.000 (Auftreten klinischer Symptome), aber auch 1:31.000 in der Slowakei und 1:19.000 bei einem Urinmassenscreening in Berlin. Im deutschen Sprachraum gehen wir von mehreren hundert Patienten aus. Ganz zweifelsfrei sind viele Patienten mit Alkaptonurie heute nicht erkannt.
Eine Behandlung mit Nitisinon ist die derzeit einzige, wirksame Therapie bei Alkaptonurie. Nitisinon hemmt das Enzym Hydroxyphenylpyruvatdioxigenase, wodurch die Bildung der Homogentisinessigsäure verhindert wird. Bisher wird Nitisinon zur Behandlung von Kindern, Jugendlichen und Erwachsenen mit der bestätigten Diagnose angeborene Tyrosinämie Typ 1 angewendet. Seit 2020 ist Nitisinon auch für die Behandlung von AKU-Patienten zugelassen. Bei Kindern im Alter von 0-18 Jahren mit Alkaptonurie ist die Sicherheit und Wirksamkeit von Nitisinon nicht erwiesen. Es liegen keine Daten vor.
Unter Nitisinoneinnahme kommt es zu einer erhöhten Konzentration von Tyrosin und Phenylalanin. Eine sehr hohe Tyrosinkonzentration kann wiederum zu einer vermehrten Kristallbildung in der Haut (z.B. Handflächen oder Fußsohlen) und in der Bindehaut der Augen führen, welche sehr schmerzhaft sein können. Um dies zu verhindern, sollte eine eiweißkontrollierte Diät eingehalten werden, um sichere Tyrosinkonzentrationen im Blut zu erreichen.
Die Einhaltung einer eiweißkontrollierten Ernährung wird erst mit Therapiebeginn von Nitisinon empfohlen. Vor Beginn der Therapie ist es nicht sicher, ob eine eiweißbewusste Diät den Krankheitsverlauf und die orthopädischen und internistischen Beschwerden oder Befunde wesentlich beeinflussen kann.
Es ist in jedem Fall zu beachten, dass dabei nicht zu wenig Eiweiß zugeführt wird. Der Körper braucht Eiweiß essenziell, zum Beispiel für den Aufbau und die Reparatur bzw. Regeneration der Muskulatur, für die Bildung von Kollagen, Immunkörpern und Enzymen, für die Sättigung und die Energielieferung.

D-A-CH Referenzwerte für die Eiweißzufuhr, Deutsche Gesellschaft für Ernährung
Quellen: Mönch, E. und Link, R., 2006, Alkaptonurie. In Mönch, E. und Link, R., Diagnostik und Therapie bei angeborenen Stoffwechselstörungen, 2. Auflage, SPS Verlagsgesellschaft.
Richter, P., Hallo, D., Le Guillou, D., Wagner, L., 2012 https://www.youtube.com/watch?v=t5EgHvSYDPM.